
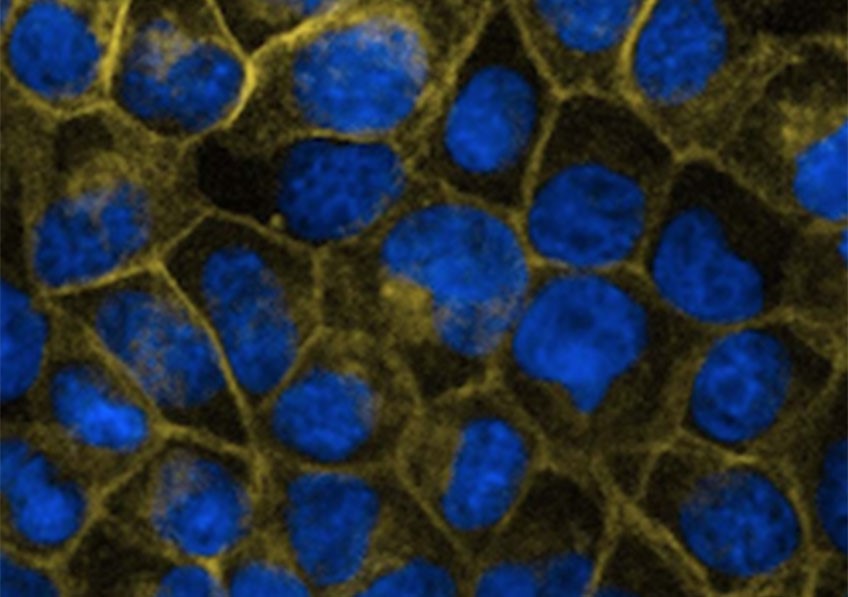
L'effet de la mutation G60R sur l'expression et la localisation de CLDN-5 dans les cellules CLDN-null. © Matthew Campbell, Smurfit Institute of Genetics, Trinity College Dublin
Des scientifiques du Trinity College de Dublin, de l’Inserm, d’Université Paris Cité et de l’AP-HP au sein de l'Institut Imagine à l'hôpital Necker, à Paris, viennent de découvrir un second gène responsable d'une maladie très rare appelée hémiplégie alternante de l'enfant (HAE). Il s'agit d'une maladie sévère qui peut entraîner une paralysie répétée affectant un côté du corps, ou l'autre, ou parfois les deux à la fois. Elle atteint généralement les enfants avant l'âge de 18 mois et, à ce jour, un seul gène responsable avait été identifié. Les résultats de ces travaux sont publiés dans la revue internationale Brain.
L'hémiplégie alternante de l'enfance (HAE) est une maladie neuro-développementale très rare qui se caractérise par une hémiplégie périodique, un retard persistant du développement et un déficit cognitif. Le signe précoce majeur est la survenue d'épisodes répétés d'hémiplégie de quelques minutes à plusieurs jours, touchant tantôt un côté du corps tantôt l'autre. Plusieurs facteurs peuvent déclencher les épisodes aigus : stress, exposition à l'eau, activités physiques, variations lumineuses et certains aliments. Il n’existe à ce jour, aucun traitement spécifique de l'HAE, qui touche moins d’un enfant sur 100 000 chaque année. La prise en charge des patients est symptomatique et pluridisciplinaire et associe des mesures prophylactiques (éviter l’exposition aux éléments déclenchants), le traitement aigu des crises le traitement de l'épilepsie et la prise en charge éducative..
Des scientifiques de Dublin et de Paris ont identifié le gène CLDN5, comme étant responsable d’hémiplégie alternante de l'enfant dans deux cas non apparentés de HAE en France. La protéine produite par ce gène, la claudine-5, est essentielle au maintien de l'intégrité de la barrière hémato-encéphalique qui isole et protège le cerveau du reste du fonctionnement de l’organisme. De manière inexpliquée, la forme mutée de la protéine transforme la barrière en un canal sélectif pour les ions chargés négativement. À cet égard, les compositions ioniques du cerveau sont probablement modifiées chez ces enfants, ce qui constitue un facteur clé de la maladie.
En plus d’apporter de nouvelles connaissances à cette maladie, ces travaux pourront avoir des répercussions sur la compréhension fondamentale de la protéine de jonction qui forme la barrière hémato-encéphalique. Comme c’est la première fois que la transformation de cette barrière humaine en un canal est mise en évidence, il pourrait y avoir des voies d'administration de médicaments qui n'ont jamais été explorées. Ces résultats serviront de base aux prochaines étapes du projet.
"Cette découverte est le fruit d'une formidable collaboration entre nos équipes françaises et irlandaises. L'identification de ces mutations de novo chez des enfants non apparentés suggérant que la barrière se transforme en canal est passionnante à plusieurs niveaux. Il s'agit de la première découverte de la transformation de la BHE en canal, mais cela éclaire également la pathologie dévastatrice de la HAE, ce qui peut aider à une meilleure la gestion clinique des patients présentant cette mutation » déclarent conjointement le Dr Matthew Campbell, professeur associé au Trinity College de Dublin et Arnold Munnich, médecin à l'AP- HP et professeur à Université Paris Cité.
Cette étude a été financée par la Science Foundation Ireland (SFI), FutureNeuro, la Société japonaise pour la promotion de la science (JSPS) et le Conseil européen de la recherche (CER).
Sources
Recurrent de novo mutations in CLDN5 induce an anion-selective blood–brain barrier and alternating hemiplegia
Yosuke Hashimoto,1,† Karine Poirier,2,† Nathalie Boddaert,3 Laurence Hubert,2 Melodie Aubart,4 Anna Kaminska,4 Marianne Alison,5 Isabelle Desguerre,4 Arnold Munnich2,4,‡ and Matthew Campbell1,‡ †
‡These authors contributed equally to this work.
1 Smurfit Institute of Genetics, Trinity College Dublin, Dublin 2, Ireland
2 INSERM UMR1163, Institut Imagine, Université Paris Cité, F-75015, Paris France
3 Department of pediatric radiology, Hospital Necker Enfants Malades, France
4 Departments of pediatric neurology and medical genetics, Hospital Necker-Enfants Malades, Université Paris Cité, F-75015, Paris France
5 Department of pediatric radiology, Hospital Robert Debré, Université Paris Cité, F-75015, Paris France
Brain https://doi.org/10.1093/brain/awac215 juin 2022